Nature ( 2026 ) Cite this article
G-protein-coupled receptor (GPCR) signalling occurs through heterotrimeric G proteins, whose selective activation leads to distinct cellular outcomes 1 .
Although more than 200 GPCR–G protein complex structures have been determined 2 , these static snapshots provide limited insight into the dynamics of G-protein association and dissociation.
Here we present cryo-electron microscopy structures of human neurotensin receptor type 1 (NTSR1) with minimally modified G o and G q , showing how the receptor’s intracellular surface dynamically rearranges to accommodate each G-protein subtype.
Furthermore, time-resolved cryo-electron microscopy analyses of NTSR1–G i visualized G-protein dissociation processes on GDP/GTP binding.
Characterization of more than 20 intermediates, complemented by mutational and computational analyses, identifies four key mechanistic features.
First, GDP/GTP induces G i release from both canonical and non-canonical active conformations with distinct kinetics.
Second, NTSR1 uses common intracellular rearrangements to recognize different G-protein subtypes and to promote activation of a single subtype.
Third, separation from Gβγ involves stepwise remodelling of the Gα switches I–III.
Finally, G i dissociates from the receptor through a pathway that is distinct from that of G s , and the canonical and non-canonical NTSR1–G i complexes further diverge in their dissociation trajectories.
These findings provide a comprehensive framework for understanding GPCR signalling dynamics and guiding signal-targeted therapeutic development.
This is a preview of subscription content, access via your institution
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
Receive 51 print issues and online access
Prices may be subject to local taxes which are calculated during checkout
Fig.
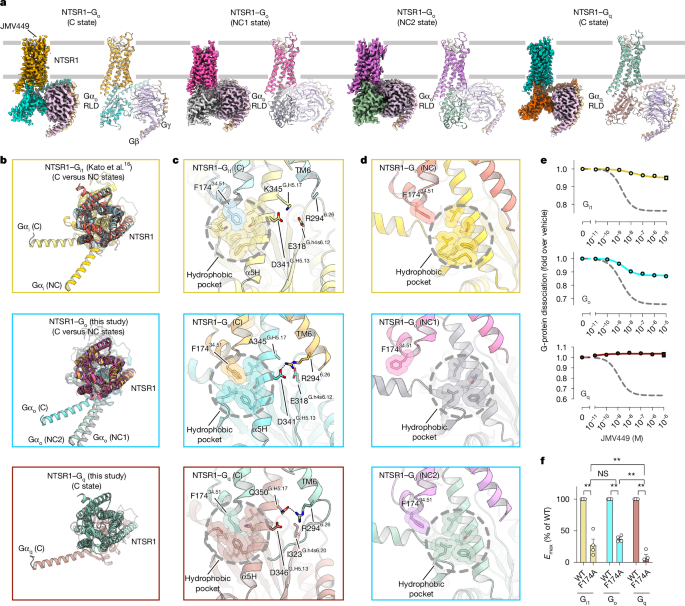
1: Cryo-EM structures of NTSR1 complexes with G o or G q in the nucleotide-free state.
Fig.
2: Time-resolved cryo-EM analysis of the NTSR1–G i complex with GDP.
Fig.
3: 3DVA captures stepwise GDP-induced G-protein dissociation from the receptor.
Fig.
4: Time-resolved cryo-EM analysis of the NTSR1–G i complex with GTP.
Fig.
5: GTP-induced G-protein dissociation in the NC state.
Similar content being viewed by others
Snapshots of the dynamic basis of NTSR1 G protein subtype promiscuity
Designing allosteric modulators to change GPCR G protein subtype selectivity
Time-resolved cryo-EM of G-protein activation by a GPCR
The cryo-EM density maps and atomic coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and the PDB with accession codes EMD-64904 and 9VAT for NTSR1–G o , C (nucleotide-free, AHD-open); EMD-64905 and 9VAU for NTSR1–G o , NC1 (nucleotide-free, AHD-open); EMD-64906 and 9VAV for NTSR1–G o , NC2 (nucleotide-free, AHD-open); EMD-64907 and 9VAW for NTSR1–G q , C (nucleotide-free, AHD-open); EMD-64908 and 9VAX for NTSR1–G i GDP 8s -C1 (GDP-unbound, AHD-open); EMD-64909 and 9VAY for NTSR1–G i GDP 8s -C2 (GDP-unbound, AHD-open); EMD-64910 and 9VAZ for NTSR1–G i GDP 8s -NC1 (GDP-unbound, AHD-open); EMD-64911 and 9VB0 for NTSR1–G i GDP 8s -NC2 (GDP-unbound, AHD-open); EMD-64912 and 9VB1 for NTSR1–G i GDP 8s -NC3 (GDP-unbound, AHD-open); EMD-64913 and 9VB2 for NTSR1–G i GDP 8s -C3 (GDP-bound, AHD-closed); EMD-64914 and 9VB3 for NTSR1–G i GDP 8s -C4 (GDP-bound, AHD-closed); EMD-64915 and 9VB4 for NTSR1–G i GDP 15s -C1 (GDP-bound, AHD-closed); EMD-64916 and 9VB5 for NTSR1–G i GDP 15s -C2 (GDP-bound, AHD-closed); EMD-64917 and 9VB6 for NTSR1–G i GTP 8s -C1 (GTP-bound, AHD-open); EMD-64918 and 9VB7 for NTSR1–G i GTP 8s -C2 (GTP-bound, AHD-closed); EMD-64919 and 9VBA for NTSR1–G i GTP 8s -C3 (GTP-bound, AHD-closed); EMD-67429 and 20ZG for i GTP 0–5s -C1 (GTP-unbound, AHD-open); EMD-67430 and 20ZH for i GTP 0–5s -C2 (GTP-unbound, AHD-open); EMD-67431 and 20ZI for i GTP 0–5s -NC1 (GTP-unbound, AHD-open); EMD-67432 and 20ZJ for i GTP 0–5s -NC2 (GTP-unbound, AHD-open); EMD-67433 and 20ZK for NTSR1–G i GTP 0–5s -NC3 (GTP-unbound, AHD-open); EMD-67434 and 20ZL for NTSR1–G i GTP 0–5s -NC4 (GTP-bound, AHD-open); EMD-67426 and 20ZC for NTSR1-delipidated G i GTP 0–5s -C (GTP-unbound, AHD-open); EMD-67427 and 20ZD for NTSR1-delipidated G i GTP 0–5s -NC (GTP-unbound, AHD-open).
The raw cryo-EM images of all datasets have been deposited in the Electron Microscopy Public Image Archive (EMPIAR).
Maps and atomic coordinates for GTP 0–5s -NC5 and the 3DVA results of GDP 8s are available at Zenodo ( https://doi.org/10.5281/zenodo.15755069 and https://doi.org/10.5281/zenodo.17731629 ) 70 , 71 .
All other data are available on request from the corresponding author.
Source data are provided with this paper.
Hilger, D., Masureel, M.
& Kobilka, B.
K.
Structure and dynamics of GPCR signaling complexes.
Nat.
Struct.
Mol.
Biol.
25 , 4–12 (2018).
Article CAS PubMed PubMed Central Google Scholar
Herrera, L.
P.
T.
et al.
GPCRdb in 2025: adding odorant receptors, data mapper, structure similarity search and models of physiological ligand complexes.
Nucleic Acids Res.
53 , D425–D435 (2025).
Duan, J.
et al.
Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy.
Nat.
Commun.
11 , 4121 (2020).
Article ADS CAS PubMed PubMed Central Google Scholar
Garcia-Nafria, J., Nehme, R., Edwards, P.
C.
& Tate, C.
G.
Cryo-EM structure of the serotonin 5-HT 1B receptor coupled to heterotrimeric G o .
Nature 558 , 620–623 (2018).
Kang, Y.
et al.
Cryo-EM structure of human rhodopsin bound to an inhibitory G protein.
Nature 558 , 553–558 (2018).
Draper-Joyce, C.
J.
et al.
Structure of the adenosine-bound human adenosine A 1 receptor–G i complex.
Nature 558 , 559–563 (2018).
Article ADS CAS PubMed Google Scholar
Olsen, R.
H.
J.
& English, J.
G.
Advancements in G protein-coupled receptor biosensors to study GPCR–G protein coupling.
Br.
J.
Pharmacol.
180 , 1433–1443 (2023).
Article CAS PubMed Google Scholar
Mustain, W.
C., Rychahou, P.
G.
& Evers, B.
M.
The role of neurotensin in physiologic and pathologic processes.
Curr.
Opin.
Endocrinol.
Diabetes Obes.
18 , 75–82 (2011).
Boules, M., Li, Z., Smith, K., Fredrickson, P.
& Richelson, E.
Diverse roles of neurotensin agonists in the central nervous system.
Front.
Endocrinol.
4 , 36 (2013).
Slosky, L.
M.
et al.
beta-arrestin-biased allosteric modulator of NTSR1 selectively attenuates addictive behaviors.
Cell 181 , 1364–1379 (2020).
Thompson, K.
D.
& Leinninger, G.
M.
Influence of the neurotensin signaling system on feeding and satiety.
Neuropharmacology 275 , 110496 (2025).
Sun, D.
et al.
Molecular mechanism of the arrestin-biased agonism of neurotensin receptor 1 by an intracellular allosteric modulator.
Cell Res.
35 , 284–295 (2025).
Duan, J.
et al.
GPCR activation and GRK2 assembly by a biased intracellular agonist.
Nature 620 , 676–681 (2023).
Deluigi, M.
et al.
Complexes of the neurotensin receptor 1 with small-molecule ligands reveal structural determinants of full, partial, and inverse agonism.
Sci.
Adv.
7 , eabe5504 (2021).
Huang, W.
et al.
Structure of the neurotensin receptor 1 in complex with beta-arrestin 1.
Nature 579 , 303–308 (2020).
Kato, H.
E.
et al.
Conformational transitions of a neurotensin receptor 1–G i1 complex.
Nature 572 , 80–85 (2019).
Zhang, M.
et al.
Cryo-EM structure of an activated GPCR–G protein complex in lipid nanodiscs.
Nat.
Struct.
Mol.
Biol.
28 , 258–267 (2021).
Krumm, B.
E.
et al.
Neurotensin receptor allosterism revealed in complex with a biased allosteric modulator.
Biochemistry 62 , 1233–1248 (2023).
Ballesteros, J.
A.
& Weinstein, H.
Integrated methods for the construction of three-dimensional models and computational probing of structure–function relations in G protein-coupled receptors.
Methods Neurosci.
25 , 366–428 (1995).
Flock, T.
et al.
Universal allosteric mechanism for Gα activation by GPCRs.
Nature 524 , 173–179 (2015).
Du, Y.
et al.
Assembly of a GPCR–G protein complex.
Cell 177 , 1232–1242 (2019).
Yamada, M., Yamada, M., Watson, M.
A.
& Richelson, E.
Deletion mutation in the putative third intracellular loop of the rat neurotensin receptor abolishes polyphosphoinositide hydrolysis but not cyclic AMP formation in CHO-K1 cells.
Mol.
Pharmacol.
46 , 470–476 (1994).
Masuho, I.
et al.
Rules and mechanisms governing G protein coupling selectivity of GPCRs.
Cell Rep.
42 , 113173 (2023).
Gregorio, G.
G.
et al.
Single-molecule analysis of ligand efficacy in β 2 AR–G-protein activation.
Nature 547 , 68–73 (2017).
Papasergi-Scott, M.
M.
et al.
Time-resolved cryo-EM of G-protein activation by a GPCR.
Nature 629 , 1182–1191 (2024).
Dror, R.
O.
et al.
Signal transduction.
Structural basis for nucleotide exchange in heterotrimeric G proteins.
Science 348 , 1361–1365 (2015).
Teng, X.
et al.
Structural insights into G protein activation by D1 dopamine receptor.
Sci.
Adv.
8 , eabo4158 (2022).
Batebi, H.
et al.
Mechanistic insights into G-protein coupling with an agonist-bound G-protein-coupled receptor.
Nat.
Struct.
Mol.
Biol.
31 , 1692–1701 (2024).
Lohse, M.
J.
et al.
Kinetics of G-protein-coupled receptor signals in intact cells.
Br.
J.
Pharmacol.
153 , S125–S132 (2008).
Knight, K.
M.
et al.
Molecular annotation of G protein variants in a neurological disorder.
Cell Rep.
42 , 113578 (2023).
Wall, M.
A.
et al.
The structure of the G protein heterotrimer G i α 1 β 1 γ 2 .
Cell 83 , 1047–1058 (1995).
Coleman, D.
E.
& Sprang, S.
R.
Structure of G i α 1 ·GppNHp, autoinhibition in a G α protein–substrate complex.
J.
Biol.
Chem.
274 , 16669–16672 (1999).
Johnston, C.
A.
et al.
Structure of Gα i1 bound to a GDP-selective peptide provides insight into guanine nucleotide exchange.
Structure 13 , 1069–1080 (2005).
Knight, K.
M.
et al.
Molecular annotation of G protein variants in a neurological disorder.
Cell Rep.
42 , 113462 (2023).
Silachev, D.
et al.
Mouse models characterize GNAO1 encephalopathy as a neurodevelopmental disorder leading to motor anomalies: from a severe G203R to a milder C215Y mutation.
Acta Neuropathol.
Commun.
10 , 9 (2022).
Nakamura, K.
et al.
De novo mutations in GNAO1, encoding a Gα o subunit of heterotrimeric G proteins, cause epileptic encephalopathy.
Am.
J.
Hum.
Genet.
93 , 496–505 (2013).
Knight, K.
M.
et al.
A universal allosteric mechanism for G protein activation.
Mol.
Cell 81 , 1384–1396 (2021).
Larasati, Y.
A.
et al.
Restoration of the GTPase activity and cellular interactions of Gα(o) mutants by Zn 2+ in GNAO1 encephalopathy models.
Sci.
Adv.
8 , eabn9350 (2022).
Liu, Q.
et al.
Ligand recognition and G-protein coupling selectivity of cholecystokinin A receptor.
Nat.
Chem.
Biol.
17 , 1238–1244 (2021).
Zhou, Y.
et al.
Molecular insights into ligand recognition and G protein coupling of the neuromodulatory orphan receptor GPR139.
Cell Res.
32 , 210–213 (2022).
Coleman, D.
E.
et al.
Structures of active conformations of G iα1 and the mechanism of GTP hydrolysis.
Science 265 , 1405–1412 (1994).
Conklin, B.
R., Farfel, Z., Lustig, K.
D., Julius, D.
& Bourne, H.
R.
Substitution of three amino acids switches receptor specificity of G q α to that of G i α.
Nature 363 , 274–276 (1993).
Lomize, A.
L., Todd, S.
C.
& Pogozheva, I.
D.
Spatial arrangement of proteins in planar and curved membranes by PPM 3.0.
Protein Sci.
31 , 209–220 (2022).
Kugawa, M.
et al.
Structural insights into lipid chain-length selectivity and allosteric regulation of FFA2.
Nat.
Commun.
16 , 2809 (2025).
Maeda, S.
et al.
Development of an antibody fragment that stabilizes GPCR/G-protein complexes.
Nat.
Commun.
9 , 3712 (2018).
Article ADS PubMed PubMed Central Google Scholar
Mastronarde, D.
N.
Automated electron microscope tomography using robust prediction of specimen movements.
J.
Struct.
Biol.
152 , 36–51 (2005).
Zivanov, J.
et al.
New tools for automated high-resolution cryo-EM structure determination in RELION-3.
eLife 7 , e42166 (2018).
Article PubMed PubMed Central Google Scholar
Rohou, A.
& Grigorieff, N.
CTFFIND4: fast and accurate defocus estimation from electron micrographs.
J.
Struct.
Biol.
192 , 216–221 (2015).
Zivanov, J., Nakane, T.
& Scheres, S.
H.
W.
A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis.
IUCrJ 6 , 5–17 (2019).
Zivanov, J., Nakane, T.
& Scheres, S.
H.
W.
Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1.
IUCrJ 7 , 253–267 (2020).
Punjani, A., Rubinstein, J.
L., Fleet, D.
J.
& Brubaker, M.
A.
cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination.
Nat.
Methods 14 , 290–296 (2017).
Zivanov, J.
et al.
A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0.
eLife 11 , e83724 (2022).
Bepler, T.
et al.
Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs.
Nat.
Methods 16 , 1153–1160 (2019).
Burt, A.
et al.
An image processing pipeline for electron cryo-tomography in RELION-5.
FEBS Open Bio 14 , 1788–1804 (2024).
Xia, R.
et al.
Cryo-EM structure of the human histamine H 1 receptor/G q complex.
Nat.
Commun.
12 , 2086 (2021).
Kim, K.
et al.
Structure of a hallucinogen-activated G q -coupled 5-HT 2A serotonin receptor.
Cell 182 , 1574–1588 (2020).
Yuan, D.
et al.
Activation of the α 2B adrenoceptor by the sedative sympatholytic dexmedetomidine.
Nat.
Chem.
Biol.
16 , 507–512 (2020).
Emsley, P.
& Cowtan, K.
Coot: model-building tools for molecular graphics.
Acta Crystallogr.
D 60 , 2126–2132 (2004).
Article ADS PubMed Google Scholar
Murshudov, G.
N.
et al.
REFMAC5 for the refinement of macromolecular crystal structures.
Acta Crystallogr.
D 67 , 355–367 (2011).
Yamashita, K., Palmer, C.
M., Burnley, T.
& Murshudov, G.
N.
Cryo-EM single-particle structure refinement and map calculation using Servalcat.
Acta Crystallogr.
D 77 , 1282–1291 (2021).
Nicholls, R.
A., Long, F.
& Murshudov, G.
N.
Low-resolution refinement tools in REFMAC5.
Acta Crystallogr.
D 68 , 404–417 (2012).
Chen, V.
B.
et al.
MolProbity: all-atom structure validation for macromolecular crystallography.
Acta Crystallogr.
D 66 , 12–21 (2010).
Goddard, T.
D.
et al.
UCSF ChimeraX: meeting modern challenges in visualization and analysis.
Protein Sci.
27 , 14–25 (2018).
Inoue, A.
et al.
Illuminating G-protein-coupling selectivity of GPCRs.
Cell 177 , 1933–1947 (2019).
Sumino, A., Uchihashi, T.
& Oiki, S.
Oriented reconstitution of the full-length KcsA potassium channel in a lipid bilayer for AFM imaging.
J.
Phys.
Chem.
Lett.
8 , 785–793 (2017).
Ando, T.
et al.
A high-speed atomic force microscope for studying biological macromolecules.
Proc.
Natl Acad.
Sci.
USA 98 , 12468–12472 (2001).
Uchihashi, T., Kodera, N.
& Ando, T.
Guide to video recording of structure dynamics and dynamic processes of proteins by high-speed atomic force microscopy.
Nat.
Protoc.
7 , 1193–1206 (2012).
Sumino, A., Sumikama, T., Uchihashi, T.
& Oiki, S.
High-speed AFM reveals accelerated binding of agitoxin-2 to a K + channel by induced fit.
Sci.
Adv.
5 , eaax0495 (2019).
Sumino, A.
et al.
High-speed atomic force microscopy reveals fluctuations and dimer splitting of the N-terminal domain of GluA2 ionotropic glutamate receptor–auxiliary subunit complex.
ACS Nano.
18 , 25018–25035 (2024).
Kobayashi, K., Kato, H.
E., Kawakami, K.
& Fukuda, M.
Conformational dynamics governing G-protein recognition and activation by neurotensin receptor 1.
Zenodo https://doi.org/10.5281/zenodo.15755069 (2025).
Kobayashi, K., Kato, H.
E., Kawakami, K.
& Fukuda, M.
Conformational dynamics governing G-protein recognition and activation by neurotensin receptor 1.
Zenodo https://doi.org/10.5281/zenodo.17731629 (2026).
We thank R.
Danev and M.
Kikkawa (UTokyo) for setting up the cryo-EM infrastructure; T.
Kusakizako and Y.
Sakamaki (UTokyo) and R.
N.
Burton-Smith and K.
Murata (NIPS) for assistance with cryo-EM data collection; K.
Hasegawa and A.
Ohira (UTokyo) for administrative support; K.
Sato, S.
Nakano and Ayumi Inoue (Tohoku University) for assistance with cell-based GPCR assays; D.
Mukai (Kanazawa University) for technical assistance with HS-AFM; and J.
Janetzko (University of Colorado) for helpful discussions.
MD simulations were partially performed using computational resources at the Center for Computational Sciences, University of Tsukuba, and the Information Technology Center, The University of Tokyo.
We also acknowledge ChatGPT, a multimodal large language model created by OpenAI, for providing guidance to improve the readability of this manuscript.
Note that, after using this tool, we reviewed and edited the content as needed and take full responsibility for the content of the publication.
The electron microscopic observations were supported by Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP22ama121002, JP23ama121002, JP24ama121002, JP25ama121002 and JP25ama121005.
Calculations were partly conducted on a supercomputer at the Research Center for Computational Science in Okazaki, Japan (project: 25-IMS-C097 to T.S.).
This work was supported by JSPS KAKENHI grants (JP24K18060/JP25H02243 to K.
Kobayashi, JP23KJ0363/JP24K18286/JP25H01338 to K.
Kawakami, JP22K21351/JP23KJ1997 to S.Y., JP24H02262/JP25K09525 to M.F., JP24K01308 to T.S., JP23K23187 to A.S., JP20H03230 to A.M., JP21H04791/JP24K21281/JP25H01016 to A.I., JP19H03163/JP22H00400/JP25H01338 to H.E.K.); AMED grant (JP223fa627001 to K.
Kobayashi, JP21zf0127005 to S.Y., JP22ama121038/JP22zf0127007 to A.I., 24bm1123057h0001 to H.E.K.); JST PRESTO (JPMJPR24OF to M.F.), JST FOREST (JPMJFP224T to A.S., JPMJFR215T to A.I., JPMJFR204S to H.E.K.), JST Moonshot (JPMJMS2023 to A.I.) and JST CREST (JPMJCR21P3/JPMJCR23B1 to H.E.K.); the Astellas Foundation for Research on Metabolic Disorders (K.
Kobayashi); the Kazato Research Foundation (K.
Kobayashi); The Senri Life Science Foundation (K.
Kawakami); The Naito Foundation (K.
Kawakami); the Hirose Foundation (K.
Kawakami); The Uehara Memorial Foundation (K.
Kawakami and A.I.); The Asahi Glass Foundation (H.E.K.); and the Takeda Science Foundation (H.E.K.).
These authors contributed equally: Kazuhiro Kobayashi, Kouki Kawakami, Toshiki E.
Matsui, Shun Yokoi
Research Center for Advanced Science and Technology, The University of Tokyo, Meguro, Japan
Kazuhiro Kobayashi, Kouki Kawakami, Toshiki E.
Matsui, Masahiro Fukuda, Tomohiro J.
Narita, Keitaro Yamashita, Junki Koyanagi, Mai Kugawa, Hisako Ikeda & Hideaki E.
Kato
Department of Life Sciences, School of Arts and Sciences, The University of Tokyo, Meguro, Japan
Toshiki E.
Matsui, Junki Koyanagi & Hideaki E.
Kato
Department of Physics, School of Science and Technology, Meiji University, Kawasaki, Japan
International Institute for Integrative Sleep Medicine (WPI-IIIS), Tsukuba Institute for Advanced Research (TIAR), University of Tsukuba, Tsukuba, Japan
PRESTO, Japan Science and Technology Agency, Kawaguchi, Japan
Department of Biological Sciences, Graduate School of Science, The University of Tokyo, Bunkyo, Japan
Tomohiro J.
Narita, Mai Kugawa & Hideaki E.
Kato
Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan
Hiroki Arai, Manae Tatsumi & Asuka Inoue
Graduate School of Pharmaceutical Sciences, Kyoto University, Kyoto, Japan
Laboratory of Biomolecular Dynamics at Nanoscale, Graduate School of Biostudies, Kyoto University, Kyoto, Japan
Mai Tambo, Takashi Sumikama & Ayumi Sumino
Nano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kanazawa, Japan
Center for Living Systems Information Science (CeLiSIS), Graduate School of Biostudies, Kyoto University, Kyoto, Japan
FOREST, Japan Science and Technology Agency, Kawaguchi, Japan
Ayumi Sumino, Asuka Inoue & Hideaki E.
Kato
Department of Molecular and Cellular Physiology, Stanford University School of Medicine, Stanford, CA, USA
CREST, Japan Science and Technology Agency, Kawaguchi, Japan
Search author on: PubMed Google Scholar
K.
Kobayashi performed time-resolved cryo-EM experiments and analysed the data.
K.
Kawakami and T.J.N.
purified and prepared NTSR1–G i complex under the supervision of K.
Kobayashi.
T.E.M.
performed the cryo-EM analysis of NTSR1–G q and NTSR1–G o complexes with the help of M.F.
and J.K.
S.Y.
performed MD simulations and analysed the data under the supervision of A.M.
K.
Kawakami, H.A.
and M.
Tatsumi performed the mutational assay and analysis under the supervision of A.I.
M.
Tambo and T.S.
performed HS-AFM experiments under the supervision of A.S.
K.Y.
helped the model refinement.
H.I.
conducted cloning and mutagenesis.
M.K.
and B.K.K.
provided input on structural considerations.
K.
Kobayashi, K.
Kawakami and H.E.K.
wrote the manuscript with input from all authors.
H.E.K.
supervised all aspects of the research.
Correspondence to Hideaki E.
Kato .
B.K.K.
is a co-founder of, and consultant for, ConfometRx.
The other authors declare no competing interests.
Nature thanks Henrik Dohlman, Ramon Guixà-González, Cheng Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig.
1 Cryo-EM analysis of nucleotide-free NTSR1–G o and NTSR1–G q complexes.
a , Enlarged views of the α5 helices from three NTSR1-G protein complexes in C states.
b , c , Enlarged views of the F174 34.51 -Gα interfaces from three NTSR1-G protein complexes in C states ( b ) and NC states ( c ).
d , Cell surface expression levels for WT and F174 34.51 A mutant NTSR1.
Bars and error bars represent the mean and SEM, respectively, of three independent experiments, each performed in duplicate.
e , Two orthogonal views of aligned structures from three complexes (NTSR1–G i , G o , and G q ; all in C states).
Dashed light green, orange, and red rectangles indicate regions highlighted in ( f ), ( g ), and ( h ), respectively.
f–h , Enlarged views of ICL3 ( f ), TM2–ICL1 ( g ), and the receptor–α5 interface ( h ) from three complexes (NTSR1–G i , G o , and G q ; all in C states).
Black dashed lines represent hydrogen bonds.
Extended Data Fig.
2 HS-AFM analysis of NTSR1–G protein interaction.
a,b , Representative HS-AFM images ( a ) and height time course ( b ) of the NTSR1–G i complex in the presence of GDP.
Scale bars, 20 nm (top) and 10 nm (bottom).
In b, the red and blue boxes indicate the G-protein-bound and -unbound states, respectively.
c , Histograms of molecular height for NTSR1–G i derived from all HS-AFM measurements.
d , Correlation functions (points) and fitted curves (lines) for the dwell times of NTSR1–G i .
The correlation function for NTSR1–G i was best fitted with a quadruple-exponential decay (F(x) = 0.205e −t/0.053 + 0.175e −t/1.16 + 0.203e −t/7.05 + 0.417e −t/88.7 ; time constants: t 1 = 0.077, t 2 = 1.70, t 3 = 10.3, t 4 = 129.8 sec; R 2 = 1.000).
e , Histograms of molecular height for NTSR1–G q derived from all HS-AFM measurements.
f , Correlation functions (points) and fitted curves (lines) for the dwell times of NTSR1–G q .
NTSR1–G q was fitted with a triple-exponential decay (F(x) = 0.439e −t/0.053 + 0.175e −t/2.02 + 0.385e −t/50.1 ; time constants: t 1 = 0.077, t 2 = 2.15, t 3 = 53.3 sec; R 2 = 1.000).
g , h , Individual height time courses for all analyzed particles of NTSR1–G i (n = 31; g ) and NTSR1–G q (n = 18; h ).
Extended Data Fig.
3 Cryo-EM analysis of the NTSR1–G i complex with GDP (GDP 8s ).
a , Time-resolved cryo-EM workflow: 10 mM GDP (final concentration, 1 mM) was first applied to a grid, followed by application of purified nucleotide-free NTSR1–G i complexes.
The solution was mixed by pipetting (~1 s), incubated for 0–52 s, and then subjected to 7 s of blotting and immediate plunge-freezing (total time: 8–60 s).
b , Overlay of the density map and the structural model of the four conformers (GDP 8s -C1 to C4) focused on the GDP-binding site.
While there is no GDP density in the binding pocket of the GDP 8s -C1 and C2 states, clear GDP density is observed in the GDP 8s -C3 and -C4 states.
c , Side view of aligned structures from four conformers (GDP 8s -C1 to C4).
The eye and arrow indicate the view shown in ( d ).
Dashed green rectangle indicates the region highlighted in ( f ).
d–h , Enlarged views highlighting key regions: TM5-6 of NTSR1 ( d ), ICL3 ( e ), ICL1 ( f ), ICL2 ( g ) and the ICL2–α5 interface ( h ).
The dashed circle in ( g ) represents a potential steric clash between I344 and F174 if F174 does not change the orientation of its side chain.
i-l , Functional analysis of the three Gα i mutants (D341 G.H5.13 A, I344 G.H5.16 A, K345 G.H5.17 A) on signaling induced by NTSR1 ( i ), BLT1 ( j, k ) and MOR ( l ).
Data represent mean ± SEM of three independent experiments performed in duplicate.
** represents P < 0.01 with one-way ANOVA followed by Dunnett’s test for multiple comparison analysis with reference to WT.
ns, not significantly different between the groups.
See also the Source Data file for additional statistics and exact p- values.
Extended Data Fig.
4 Cryo-EM analysis of the NTSR1–G i complex with GDP (GDP 15s ) and population analysis across multiple incubation time points.
a , Cryo-EM density maps and structural models captured 15 s after GDP addition reveal two canonical-like conformers (GDP 15s -C1 and C2), resolved at 2.9 and 3.1 Å, respectively.
b , Time-dependent population distribution of the conformational states.
The percentage of particles assigned to each non-decoy class is plotted for the 8, 15 and 60 s datasets.
Hatched boxes indicate that an interpretable cryo-EM map could not be reconstituted using the allocated particles.
c , Overview of the image-processing workflow.
Particles extracted from the 8, 15 and 60 s datasets were pooled and subjected to heterogeneous refinement using the indicated reference maps (“ref.”).
Maps outlined in grey failed to yield interpretable structures, whereas maps outlined in red represent successfully reconstructed maps.
Extended Data Fig.
5 3DVA of the GDP 8s NTSR1–G i complex.
a , Data-processing workflow of 3DVA (top) and local resolution maps (bottom) for the GDP 8s NTSR1–G i complex.
Extended Data Fig.
6 MD simulations comparing GTP binding stability starting from deep versus shallow binding positions.
a , b , Time courses of distances from 1-µs MD simulations (n = 10 independent runs) initiated from two distinct starting configurations: a modeled structure based on GTP 8s -C1 with GTP artificially placed in the deep binding pocket ( a , Products 1–10), and the GTP 8s -C1 structure with GTP naturally located in the shallow binding pocket ( b , Products 11–20).
Blue traces represent the distance between the Cα atom of K46 G.H1.01 and the O3A atom of the GTP phosphate group.
Green traces represent the distance between the Cα atom of N269 G.S5.07 and the C5 atom of the GTP guanine ring.
c , d , Probability density histograms of the N269 G.S5.07 –GTP (guanine) distance derived from the ten simulations in a ( c , Products 1–10) and b ( d , Products 11–20).
Extended Data Fig.
7 Step-by-step comparison of switch I-III regions.
a–e , Structural comparisons of the switch I (Swi I) (top), switch II (Swi II) (middle), and switch III (Swi III) (bottom) regions between GDP-bound inactive Gα i (PDB: 1GP2 ) and GDP 8s -C1 ( a ), GDP 8s -C1 and GTP 8s -C1 ( b ), GTP 8s -C1 and GTP 8s -C2 ( c ), GTP 8s -C2 and GTP 8s -C3 ( d ), and GTP 8s -C3 and GppNHp-bound active Gα i (PDB: 1CIP ) ( e ).
Extended Data Fig.
8 Stepwise GTP-induced conformational changes in switch regions during G-protein dissociation.
a–e , Structural comparisons of switch I-III (Swi I-III) regions between GDP-bound inactive Gαi (PDB: 1GP2 ) and GDP 8s -C1 ( a ), GDP 8s -C1 and GTP 8s -C1 ( b ), GTP 8s -C1 and GTP 8s -C2 ( c ), GTP 8s -C2 and GTP 8s -C3 ( d ), and GTP 8s -C3 and GppNHp-bound active Gαi (PDB: 1CIP ) ( e ).
The black dashed circle represents the potential steric clash between the switch I loop in the GTP 8s -C2 state and the switch II-β3 loop in the GTP 8s -C1 state.
f–k , Interactions between the switch II-β3 loop and the switch III loop.
Both loops are ordered but do not strongly interact with each other in GDP-bound inactive Gαi ( f ).
When the GDP is released, the switch III loop becomes disordered ( g ), and subsequently, when GTP binds, the switch III loop partially orders ( h ).
The AHD closure brings these two loops close together ( i ), and the interaction between them, including R205 and D237, pulls out the switch II helix ( j ).
The rearranged structure of the switch II helix is further stabilized by multiple salt bridges ( k ).
Black dashed lines represent salt bridges.
Extended Data Fig.
9 Characterization of GTP 0-5s structures.
a , Structural models and local resolution maps of G-protein regions in the GTP 0-5s -NC4 (top) and GTP 0-5s -NC5 states (bottom).
The local resolutions of Gα, especially around the nucleotide-binding pocket, are markedly worse than in the receptor region.
b , Superimposed images of the cryo-EM density map and model for the GTP 0-5s -NC4/5 and GTP 8s -C3 structures.
AHD density is clearly visible next to Gβγ in both GTP 0-5s -NC4/5 states, indicating that AHD is not closed in either structure.
c , Superimposed images of the cryo-EM density map and model for the free G-protein structures, observed under the GTP 0-5s conditions.
While the left Gαi structure, which shows a closed AHD and a withdrawn α5 helix, could be reconstituted from other samples as well, the right Gαi structure, characterized by an open AHD and an extended α5 helix, could only be reconstituted from this sample.
This suggests that Gαi can be dissociated from the receptor without closing AHD and withdrawing the α5 helix at an early reaction stage (0–5 s), supporting the idea that the Gi dissociation pathway from the NC state is significantly different from that from the C state.
Extended Data Fig.
10 Working model of dynamic heterotrimeric G i protein association and dissociation processes mediated by NTSR1.
Overall association and dissociation processes mediated by NTSR1.
The processes are categorized into three primary steps: (1) association of GDP-bound G protein with the receptor (dashed orange box), (2) formation of a nucleotide-free state upon GDP release (dashed magenta box), and (3) dissociation of GTP-bound G protein from the receptor and heterotrimer separation (dashed green box).
Note that dissociation process from the NC state proceeds through a pathway distinct from that of the C state.
Both processes were analyzed starting from the most stable nucleotide-free state, with backward and forward reactions triggered by GDP and GTP, respectively.
Extended Data Fig.
11 Structural comparison of NTSR1–G i and β2AR–G s complexes obtained by time-resolved cryo-EM analysis.
a,b , Comparison of frames 1 and 20 from the NTSR1–G i structures extracted from 3DVA of the 8-s GDP dataset ( a ) and the GDP-bound inactive G i structure (PDB: 1GP2 ) ( b ), highlighting the α1 and α5 helices.
c , d , Comparison of frames 1 and 20 from the β2AR–G s structures obtained from 3DVA (PDB: 8UNL and 8UO4 , respectively) ( c ) and the GDP-bound inactive G s structure (PDB: 1BH2 ) ( d ), highlighting the α1 and α5 helices.
e , f , Interaction between the α5 helix and the β2-β3 loop from frame 1 of NTSR1–G i ( e ) and frame 1 of β2AR–G s ( f ) complexes.
The interaction is stronger in the β2AR–G s complex.
g , Comparison of frame 1 of the NTSR1–G i and β2AR–G s structures, highlighting the angle of the α5 helix.
h , Computational analysis of the potential steric clash between the α1 and α5 helices in the β2AR–G s complex.
The α5 helix from frame 1 is computationally superimposed onto the position of the α5 helix in frame 20, while preserving its original orientation.
This reveals a notable clash, suggesting that the α5 helix must be unwound and pulled out via a curved trajectory to avoid the steric clash.
The dashed circle indicates a steric clash between the α5 helix from frame 1 and the αN helix.
i , j , Comparison of α5-helix conformations between G i ( i ) and G s ( j ) in GDP-bound inactive structures.
While Gi maintains the helical structure except for the C-terminus, G s has an unwound and kinked structure near C379 G.H5.11 .
Supplementary Information (download PDF )
Supplementary Figs.
1–20, Tables 1 and 2 and legends for Supplementary Videos 1–4.
Reporting Summary (download PDF )
Peer Review File (download PDF )
Supplementary Video 1 (download MP4 )
Representative HS-AFM video of NTSR1-G i complex.
HS-AFM video of NTSR1 reconstituted in a lipid bilayer under equilibrium buffer conditions (20 mM HEPES-KOH pH 7.5, 50 mM KCl, 1 µM JMV449, 20 µM GDP, containing 200 nM delipidated Gi).
Supplementary Video 2 (download MP4 )
Representative HS-AFM video of NTSR1-only.
HS-AFM video of NTSR1 reconstituted in a lipid bilayer under equilibrium buffer conditions (20 mM HEPES-KOH pH 7.5, 50 mM KCl, 1 µM JMV449, 20 µM GDP).
Supplementary Video 3 (download MP4 )
Representative HS-AFM video of NTSR1–G q complex.
HS-AFM video of NTSR1 reconstituted in a lipid bilayer under equilibrium buffer conditions (20 mM HEPES-KOH pH 7.5, 50 mM KCl, 1 µM JMV449, 20 µM GDP, containing 500 nM delipidated Gq).
Supplementary Video 4 (download MP4 )
Structural transition illustrating opening and closing movements of the AHD, and pull-in and pull-out movements of the α5 helix.
An integrated video from 20-frame maps of the 3DVA trajectory shown in Extended Data Fig.
5 and Supplementary Fig.
13.
Source Data Figs.
1, 2 and 4, Extended Data Figs.
1–4 and 6, and Supplementary Fig.
14.
(download XLSX )
Springer Nature or its licensor (e.g.
a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Version of record : 11 March 2026
DOI : https://doi.org/10.1038/s41586-026-10228-w
Related Stories



Source: This article was originally published by Nature News
Read Full Original Article →
Comments (0)
No comments yet. Be the first to comment!
Leave a Comment